In this guide, I will give you some of my thoughts on
Additional information on qPCR primer design can be found in our Mastering qPCR course. Use code 20QPCR to get 20% off!
These features are mainly applicable to qPCR primers using intercalating dye methods (eg SYBR Green) for gene expression analyses, however, some will be translatable to other types of qPCR.
PCR product size: 70–200bp
One of the main features of a qPCR primer pair to look out for is the size of the product that will be created. It is recommended to design primers which will produce a product of 70–200bp.
Too large a product size (>200bp) can reduce the primer efficiency of the qPCR primers, whereas too small a product size (70bp) can make it difficult to differentiate them from contaminating primer-dimer formation.
I recommend aiming for a product size of around 150bp in size.
Primer length: 18–22bp
Another important feature of a qPCR primer pair is the length of the primers. Ideally, each primer should have a length of 18–22bps; usually 20bp is a common choice.
Too small a qPCR primer (<18bp) can increase the likelihood that it will bind to elsewhere in the genome, ie it is not specific enough to the target of interest. Otherwise, too large a qPCR primer (>22bp) can raise the primer melting temperature (Tm) which will impact on the annealing temperature and primer binding properties of the reaction.
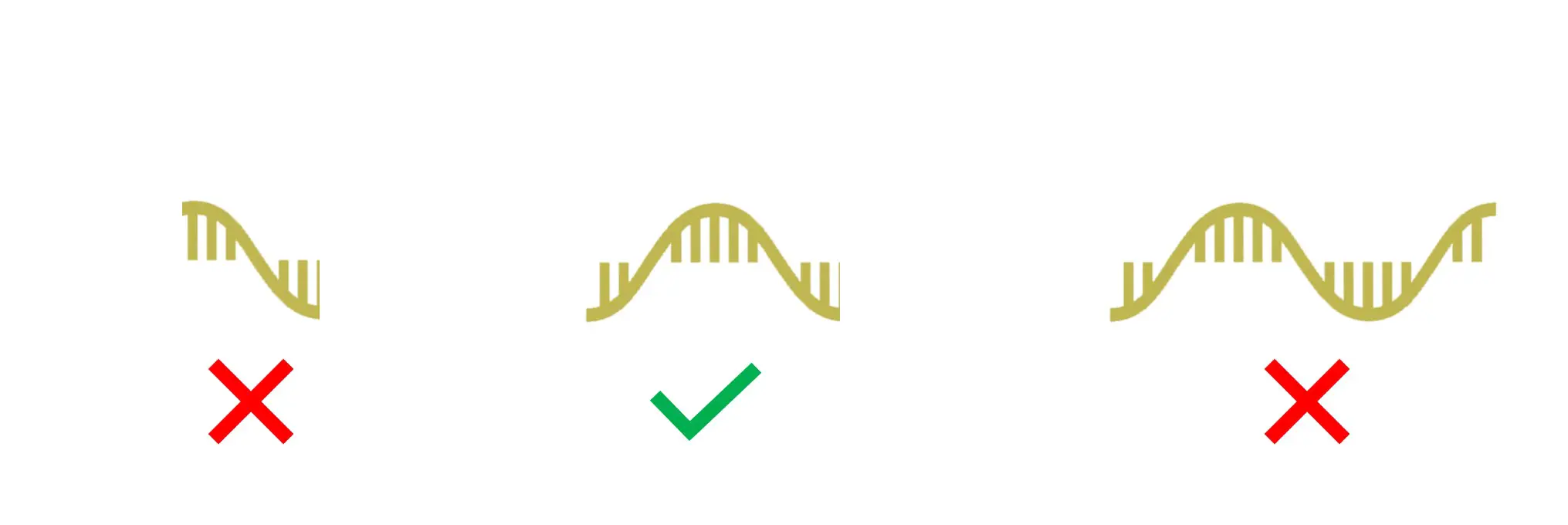
Tm: 59–65oC
The Tm is another important feature of a good qPCR primer pair. In this regard, the Tm is the temperature at which 50% of the primer is dissociated to become single stranded. Both qPCR primers should have a Tm between 59–65oC and be within 1oC of each other (no more than >3oC difference).
The Tm also relates to the annealing temperature of the reaction. As a rough rule of thumb, the annealing temperature is approximately 5oC lower than the Tm of the primers. This is only an estimation, however, since there are other parameters to consider in order to accurately deduce the annealing temperature.
Primers contain a GC clamp
Another feature which I like to include in both my primers is a GC clamp. Simply, a GC clamp is the presence of either a guanine (G) or cytosine (C) base in the last 5 bases of the primer.
The reasoning behind using a GC clamp in primers is the fact that G and C bases contain stronger hydrogen bonds, compare with adenine (A) and thymine (T) bases. Therefore, by including at least one G or C base towards the end of the primer will ensure it binds completely to the template sequence.

Be careful, however, not to include too many G or C bases in the clamp, since this can result in a reduction in specificity.
GC content: 50–60%
The GC content is simply the amount of G and C bases present in the primer sequence. It is recommended that this is kept at 50–60%; both primers should have similar values.
Having too high a GC content can result in mispriming, since GC bonds have a higher affinity to the template, compared with AT bases, they are more attracted to bind to one another.
Avoid nucleotide repeats
When analysing potential candidate qPCR primers, pay particular attention to any nucleotide repeats in the sequence. Look out for stretches of >3 of the same nucleotides in a row. These repeats can reduce the specificity of the primer and can result in mispriming.

Primers separated by an intron
One of the most important features of a good qPCR primer pair, I believe, is to design the forward and reverse primers to bind to separate exons on the gene. By doing so, this will limit the chance of genomic DNA amplification during the reaction.
DNA contains introns, whereas these are no longer present in mRNA transcripts. Thus, by separating the primers by an intron can reduce the likelihood of genomic DNA amplification, especially if the intron is very large.
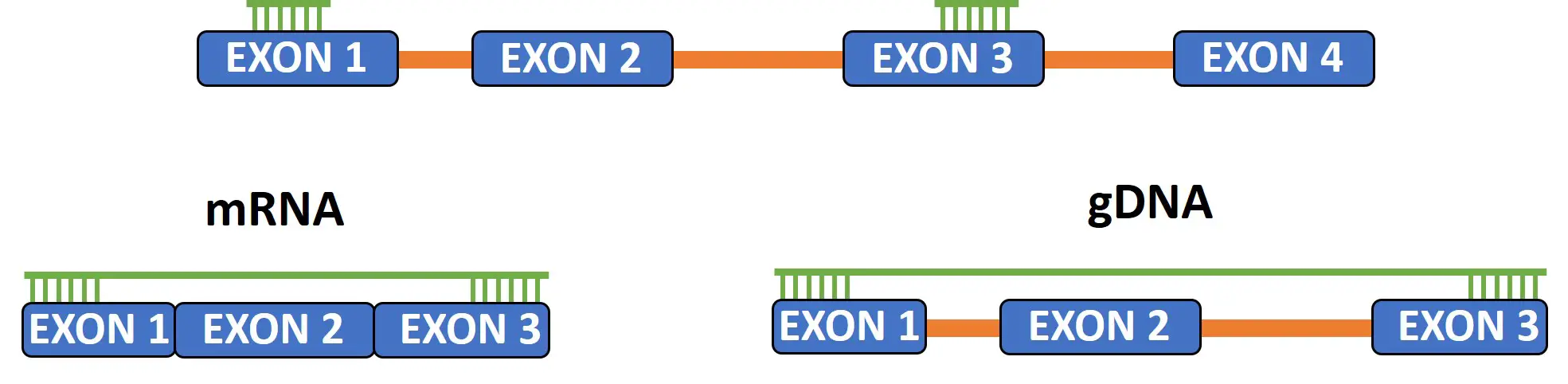
Focus on the positioning of the qPCR primers
Another aspect to consider when designing qPCR primers is the location of the primer binding along the gene of interest, especially when targetting cDNA transcripts.
For cDNA which has been created using random hexamers, is it advisable to design the primer pairs to bind near the start of the gene (5′ end).
On the other hand, for cDNA which has been created using oligo(dT) primers, design the primers to bind at the 3′ end of the gene. Since oligo(dT) primers target the polyA tail of mRNA transcripts, most of the cDNA template will be
The primer pair is specific to the target of interest
This may seem like an obvious feature, but it is one of the most important ones. Primer design software such as NCBI’s Primer-BLAST can identify any potential mispriming to other parts of the genome. This is the reason why Primer-BLAST is my favourite primer design tool.
When running Primer-BLAST, pay particular attention to any products on potentially unintended templates and their product length under each candidate primer pair. If the product length is the same as your product of interest, then re-design the primers
Also, be sure to check out our guide on designing qPCR primers with Primer-BLAST.
Conclusion
In this guide, I have discussed some of the good features of a qPCR primer pair. By adding these features into your primers during primer design, you should be able to increase your reaction efficiency.



Your blog is so helpful, it’s like a senior scientist holding your hands in the lab. Keep it up. I learnt alot from some of your write-ups
Thanks for the feedback Okoye!
what about the divergent primer for circular RNA ?
Hi Mona,
Many thanks for your suggestion. You are correct, divergent primers, which span the backsplice junction of circular RNA, are useful when detecting circular RNA.
This article mainly concerns mRNA detection.
Thanks again,
Steven
thanks steven , god bless you 🙂